Why is it important to know about immunotherapies?
- One of the largest non-governmental entities in healthcare space are Pharma and Biotech companies
- Most of the top Pharma and Biotech companies (certainly top 10) generate bulk of their revenue from oncology (largest therapeutic area) and invest heavily in the same space
- In clinical trials the largest therapeutic area is oncology (number or trials or pipeline or investment)
- The therapies in oncology (loosely speaking) has evolved from surgery, chemotherapy, radiation therapy, hormonal therapy, stem cell transplant to immunotherapy
- Immunotherapies offer significant benefits in cancer patients and are increasingly becoming part of standard of care (NCCN guidelines)
- Thus immunotherapies are focal point for many stakeholders - patients, regulators, drug developers, payers etc.
- As a result if you work for a company building solutions for life-science organisations, it is unlikely to miss Immunotherapies
- Note: Although Immunotherapies are primarily used to treat cancer, they can be used to treat other conditions as well.
What is immunotherapy?
A type of therapy that uses substances to stimulate or suppress the immune system to help the body fight cancer, infection, and other diseases. Some types of immunotherapy only target certain cells of the immune system. Others affect the immune system in a general way. - NCI
What kind of therapies are considered as immunotherapies?
A bit outdated source but still useful. The landscape has evolved significantly since this.

source: Cancer Immunotherapy

source: cancersupportcommunity.org
The National Cancer Institute has a clarification system which is uptodate but not that different.
- Immune checkpoint inhibitors, which are drugs that block immune checkpoints. These checkpoints are a normal part of the immune system and keep immune responses from being too strong. By blocking them, these drugs allow immune cells to respond more strongly to cancer.
Learn more about immune checkpoint inhibitors.
- T-cell transfer therapy, which is a treatment that boosts the natural ability of your T cells to fight cancer. In this treatment, immune cells are taken from your tumor. Those that are most active against your cancer are selected or changed in the lab to better attack your cancer cells, grown in large batches, and put back into your body through a needle in a vein.
T-cell transfer therapy may also be called adoptive cell therapy, adoptive immunotherapy, or immune cell therapy.
Learn more about T-cell transfer therapy.
- Monoclonal antibodies, which are immune system proteins created in the lab that are designed to bind to specific targets on cancer cells. Some monoclonal antibodies mark cancer cells so that they will be better seen and destroyed by the immune system. Such monoclonal antibodies are a type of immunotherapy.
Monoclonal antibodies may also be called therapeutic antibodies.
Learn more about monoclonal antibodies.
- Treatment vaccines, which work against cancer by boosting your immune system’s response to cancer cells. Treatment vaccines are different from the ones that help prevent disease.
Learn more about cancer treatment vaccines.
- Immune system modulators, which enhance the body’s immune response against cancer. Some of these agents affect specific parts of the immune system, whereas others affect the immune system in a more general way.
Learn more about immune system modulators.
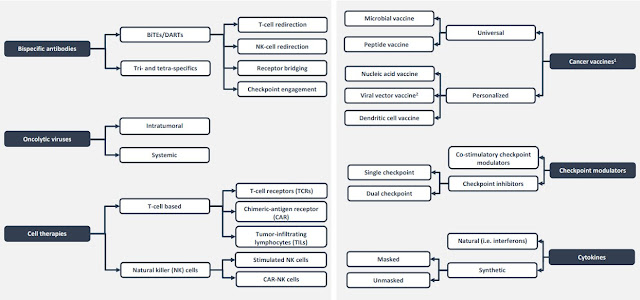
Another recent classification shows key classes within Immunotherapies below (source unknown unfortunately)