The regulators seem to consider a medical Software as a device provided it meets certain criteria (trying to diagnose, treat or enables clinical decision making etc).
There are three influential bodies when it comes to Medical Device Software matters.
The FDA, the EMA, and the International Medical Device Regulators Forum (IMDRF)
1. The 30,000 feet view:

Source: Yale
Note: The FDA will not actually run your software but rely on your documentation.
2. Qualifying as a Medical device
There are three influential bodies when it comes to Medical Device Software matters.
The FDA, the EMA, and the International Medical Device Regulators Forum (IMDRF)
2.1 IMDRF and SaMD
2.2 FDA and SaMD (Software as Medical Device)
SaMD definition: “Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”
More about IMDRF:
The FDA refers to the IMDRF definition and does not have its own separate definition.
In Europe (MDCG 2019-11), MDSW is defined as:
“Medical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.”
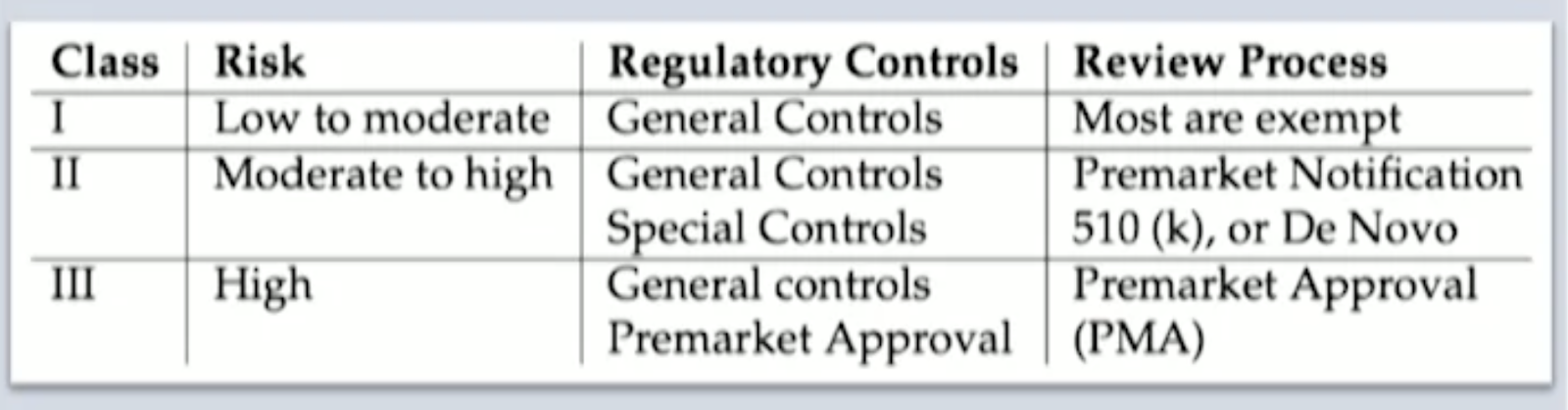
3. Class of Medical Device Software
3.1 The FDA: Here again, the FDA borrows a framework for risk categorization from the IMDRF, as noted on the FDA website.

source:imdrf.org, page 14
For SaMD pathway
3.2 For EMA

The key standards in medical software are listed below
- ISO 13485: this is the quality management systems description for medical devices.
- ISO 49721: this is the application of risk management to medical devices. These two are known as so-called management standards.
- IEC 62304: this is the system lifecycle process.
- IEC 62366: this is usability engineering, how we design software to avoid user errors and keep them safe.
These are what some SMEs call a soft laws. They do not have the force of law, but the regulatory agencies look at compliance with the standards if you follow the procedures laid out in the standards as evidence of good work, and the standards are recognized.
You have a much easier time with many regulatory policies. Well, our software process follows IEC 62304, they just know exactly what you're doing and they're very happy when you say something like that. The standards may not be 100 percent in fulfilling the needs of a particular jurisdiction. ISO 13485, is not one-for-one, the same as what the FDA would look for in an equivalent system, but it's close and it gets you most of the way there.
5. The Quality Management
You have a much easier time with many regulatory policies. Well, our software process follows IEC 62304, they just know exactly what you're doing and they're very happy when you say something like that. The standards may not be 100 percent in fulfilling the needs of a particular jurisdiction. ISO 13485, is not one-for-one, the same as what the FDA would look for in an equivalent system, but it's close and it gets you most of the way there.
5. The Quality Management
FDA’s guidance is 7 pages long
Requires signed and dated review documents for design (and also other aspects).
Subpart C: one of the 10 sections, and very important as it is for design controls.
- There are 10 sections
- Contains very high-level instructions
- Also called CFR part 810
- Also called 2017/745 MDR
- 225 pages long
- Also contains 10 sections
Requires signed and dated review documents for design (and also other aspects).
Subpart C: one of the 10 sections, and very important as it is for design controls.

source: fda.gov
6. General Principles of Software Validation
(Also called GPSV)
The scope of this guidance is somewhat broader than the scope of validation in the strictest definition of the term the way with defining. Planning, verification, testing, traceability, configuration management, and many other aspects of good software engineering discussed in this guidance are important activities that together help to support a final conclusion that the software is validated.
The updates are a very significant part of the software lifecycle, and updates can be a big source of trouble. The maintenance process is treated with the same level of seriousness as
the original process of creating the software.

The section 5 of the GPSV talks about tasks and activities in detail:

The Trifecta for FDA approval
Fundamentally the FDA wants to ensure three things,- Efficacy: does it work effectively and performs as per the claims
- Safety: it does not cause harm to the patient or caregiver (usability)
- Security: sign to prevent malicious use (cybersecurity)
7. SaMD FDA approval pathways
We have three different review processes for the different types of software.1. 510k or PMN2. De Novo3. PMA
The most common one in software, I'm sure many of you have heard the expression 510k. Here the FDA determines that the device, the software and tell me other what device you can insert the word software in your mind is substantially equivalent and has similar safety profile to a previously commercialized product. This is called the predicate.
In this particular case, if you have a piece of software that does something, for example, detect tumors from MRI images in this a previous piece of software, different products somebody else's marketed that does more or less the same thing, you can claim that that is your predicate
and all you have to do is say, we're similar to something you have already approved.This is a 510k process. A lot of time in this business is spent looking for the predicate because it makes life simpler. In the cases where there is not a predicate, but we're still in class 1 or class 2, this is type 1 type 2 classification, there's something called the De Novo process.
Is a newer process here. We'll talk a little bit about more detailed in a video clip in a minute and your device here is reviewed on its own, but you have to provide evidence of safety and effectiveness for the intended use. There is no predicate here so you say,
Pre-market Approval.
This is where we must demonstrate reasonable assurance of safety and effectiveness. This is really complicated, expensive, and your company has to be set up with a full college system as if your product was already on the market.
Approximate distribution on the 3 classess:

No comments:
Post a Comment